
In the last post, I showed you that type 2 diabetes is not a disease that starts when your fasting glucose crosses 100. It’s a disease that’s been running silently in your body for somewhere between fifteen and twenty-four years before that moment, and the standard screening tests have been missing the entire upstream phase.
This post answers the question that one raises: what is actually causing all of this? What is the underlying mechanism that drives the hyperinsulinemia, the ectopic fat accumulation, the beta cell stress, the cardiovascular damage, the brain insulin resistance, the cascade of complications that defines modern type 2 diabetes?
The answer is something the medical literature has been quietly settling on since 2008 — but that has somehow not made it into the patient conversation in any meaningful way. It is called the Twin Cycle Hypothesis, and it was first proposed by a British researcher named Roy Taylor at Newcastle University. Since then, it has been confirmed through three landmark intervention trials, dozens of MRI imaging studies, and a body of experimental work that is essentially settled science.
If you understand the Twin Cycle, you understand type 2 diabetes — what causes it, what reverses it, and why your doctor’s prescription pad has been treating the wrong target.
Let me walk you through it.
The personal fat threshold
The framework starts with a concept that completely rewrites how we should think about body composition and metabolic disease: every person has a personal fat threshold.
The personal fat threshold is the maximum amount of fat your subcutaneous tissue — the fat layer directly under your skin — can safely store. Subcutaneous fat is the body’s designed-for-purpose fat storage system. It is metabolically passive. It sits where it’s supposed to sit. It does not, under normal conditions, cause any disease.
The threshold varies enormously by individual. Some people can store 100 pounds of fat subcutaneously without metabolic consequence. Others run into trouble at 15 extra pounds. The variable is genetic, partly hormonal, and influenced by stress, sleep, age, and ethnicity.
If you want to know whether you’re over your personal fat threshold, BMI is the wrong tool. BMI was developed in the 1830s as a population statistic, not an individual clinical measure. It does not distinguish muscle from fat. It ignores fat distribution entirely. It produces dramatically different metabolic risk profiles for people with identical BMI values, and it uses the same cutoffs across populations whose genetics make those cutoffs meaningless. A lean, muscular athlete can be flagged as “overweight” by BMI. A normal-looking person with full type 2 diabetes can be flagged as “healthy weight.” The metric is broken.
The better anthropometric — increasingly endorsed by international metabolic guidelines — is waist-to-height ratio (WHtR). The rule is simple: your waist measurement at the navel should be less than half your height. Under 0.50 is generally healthy. 0.50 to 0.59 indicates elevated metabolic risk. 0.60 or above is high risk. WHtR works across all ethnicities (BMI doesn’t — South Asian populations develop metabolic disease at far lower BMIs than the cutoffs assume), captures the central obesity pattern that drives the Twin Cycle directly, and outperforms BMI for predicting type 2 diabetes, cardiovascular events, and all-cause mortality across multiple peer-reviewed studies.
But the cleanest answer is to skip anthropometrics entirely and look at the actual metabolic markers. Fasting insulin under 10 (ideally under 5). Trig/HDL ratio under 1.5. HbA1c under 5.7%. hsCRP under 1.0. ApoB under 80. If those five markers are clean, you are metabolically healthy regardless of what your body looks like on the outside. If they’re not, you have metabolic dysfunction regardless of how lean you appear. The blood panel bypasses the body composition debate entirely. Body shape is a screening signal. Blood work is the verdict.
What matters is what happens when you exceed the threshold.
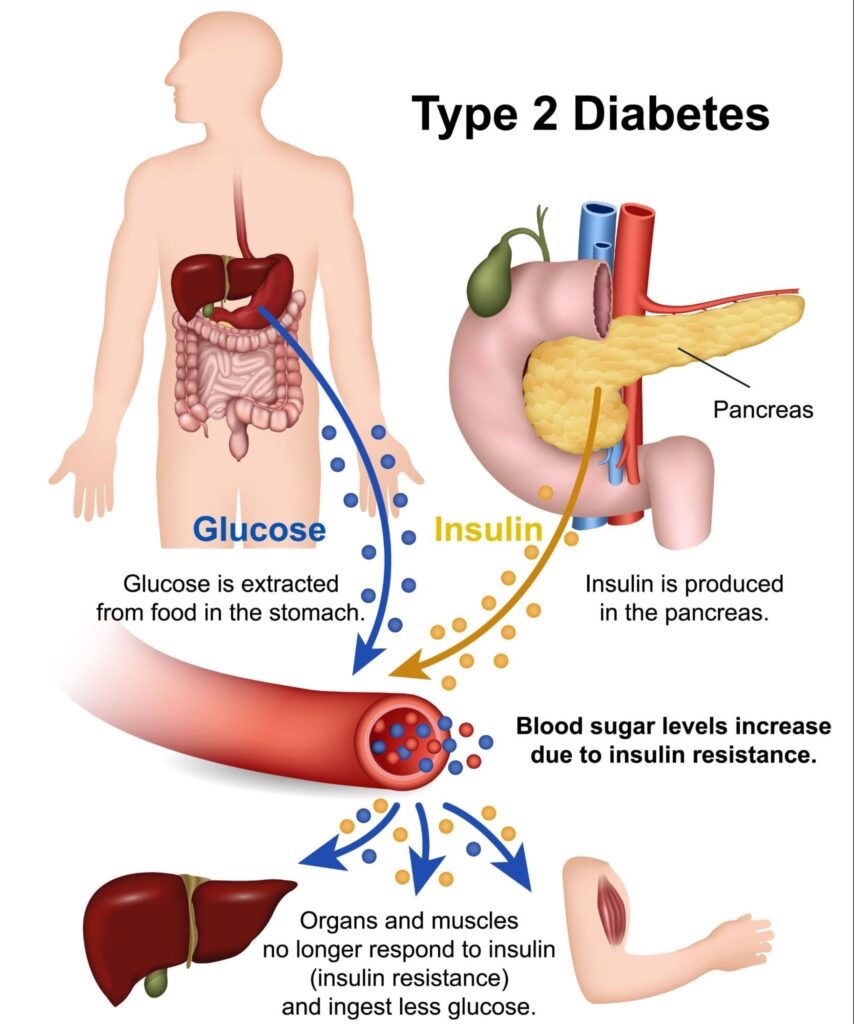
When subcutaneous storage capacity is reached, the body keeps consuming the same number of calories that put it there. The energy has to go somewhere. So it starts depositing fat ectopically — outside the fat tissue where it belongs. The liver is the first major destination. The pancreas is the second. The heart, the kidneys, the visceral cavity around the gut, even the muscle tissue itself — all of these can accumulate fat once the subcutaneous compartment is overloaded.
This is the start of the disease. Not weight gain in itself, but ectopic fat deposition in organs that were never designed to store it.
Cycle One: the liver
When fat starts to accumulate in the liver, the liver’s response to insulin starts to change. The liver is one of the body’s primary glucose-regulating organs. Under normal conditions, when insulin rises, the liver stops producing glucose (a process called gluconeogenesis) and instead starts storing it. The system is responsive, dynamic, and tightly regulated.
A fatty liver does not respond properly to insulin. The liver becomes insulin resistant at the hepatic level. Even when insulin is signaling “stop producing glucose,” the liver keeps producing glucose. Blood glucose rises slightly. The pancreas releases more insulin to compensate. The fatty liver keeps overproducing glucose anyway.
But the liver isn’t only producing glucose. It’s also producing fat. Specifically, it’s packaging triglycerides into VLDL particles — very-low-density lipoproteins — and shipping them out into the bloodstream. This is the same VLDL pathway that drives the lipid abnormalities I walked through in the cholesterol series. High triglycerides, low HDL, small dense LDL, atherogenic dyslipidemia — these are not separate diseases from type 2 diabetes. They are the cardiovascular manifestation of the same liver pathology.
The VLDL particles travel through the bloodstream looking for somewhere to deliver their triglyceride cargo. Some of it goes to fat tissue, where it deepens the original storage problem. Some of it deposits in arterial walls, where it contributes to atherosclerotic plaque. And some of it ends up at the pancreas.
This is the bridge from Cycle One to Cycle Two.
Cycle Two: the pancreas
The pancreas houses the beta cells, the specialized cells that produce insulin in response to blood glucose. Beta cells are sensitive structures. They function properly only under specific metabolic conditions. One of those conditions is that they should not be coated in fat.
When VLDL particles from the fatty liver start delivering triglycerides to the pancreas, beta cells begin to accumulate fat. This is called pancreatic steatosis, and it has direct consequences for insulin production. Roy Taylor’s research showed through high-resolution MRI imaging that pancreatic fat percentage in type 2 diabetics is measurably elevated compared to healthy controls — and that this fat directly impairs beta cell function.
In Taylor’s own words: “We saw that when a person accumulates too much fat, which should be stored under the skin, then it has to go elsewhere in the body. The amount that can be stored under the skin varies from person to person, indicating a ‘personal fat threshold’ above which fat can cause mischief. When fat cannot be safely stored under the skin, it is then stored inside the liver, and over-spills to the rest of the body including the pancreas.”
The metaphor Taylor uses is “clogging up” the pancreas. The fat physically interferes with the machinery of insulin production. The genes responsible for the beta cell phenotype start switching off. Insulin production drops. Glucose rises. The disease becomes manifest.
But here is the critical part: this entire cascade is reversible. Remove the dietary input that’s driving the liver fat accumulation, force the body to mobilize ectopic fat back out of the liver and pancreas, and the beta cells — as long as enough of them are still alive — will resume normal function. The pancreas can come back online. Glucose normalizes. The disease resolves.
This is not theoretical. This is what the DiRECT and ReTUNE trials demonstrated, and what hundreds of patients have reproduced under medical supervision. We’ll cover that data in detail in Post 4.
The dietary driver: fructose specifically
The Twin Cycle gives us the mechanism. The next question is: what’s actually putting all that fat into the liver in the first place?
The answer is more specific than most people realize. The dietary driver of liver fat accumulation is not generic “calories.” It is not even generic “carbohydrates.” It is fructose — and the precision matters enormously.
Glucose and fructose are both sugars, both six-carbon molecules, and both contain four calories per gram. Their fates in the body are radically different. When glucose is consumed, it enters the bloodstream, where it can be used by any cell in the body. Insulin is released to facilitate uptake. The liver takes its share, but most of it is used directly by muscle tissue, brain tissue, and other consuming organs. Glucose has multiple regulatory checkpoints along its metabolic pathway, and the body is sophisticated about managing it.
Fructose has almost none of those checkpoints. Fructose bypasses key regulatory steps in glycolysis, speeding liver fat synthesis. Roughly 90% of consumed fructose is metabolized in the liver, where it is converted directly into fat through a process called de novo lipogenesis. Excess liver fat impairs insulin clearance and promotes systemic insulin resistance.
The peer-reviewed research has now made this clear: “The current evidence shows that the fructose, but not glucose, component of dietary sugar drives metabolic complications and contradicts the notion that fructose is merely a source of palatable calories that leads to increased weight gain and insulin resistance.” Fructose is not just sugar. It is specifically the sugar that drives the disease.
Where does fructose come from in the modern diet?
- Table sugar (sucrose) — 50% fructose, 50% glucose
- High-fructose corn syrup — typically 55% fructose, 42% glucose
- Agave nectar — up to 90% fructose
- Fruit juice concentrate — concentrated fructose load, no fiber
- Honey — about 40% fructose
- Whole fruit — contains fructose, but in a form packaged with fiber, water, and slow-release matrix that limits liver impact dramatically
Whole fruit is not a meaningful contributor to the disease. The fiber slows fructose absorption, the water content is limiting, and the volume required to eat enough whole apples to get a problematic fructose dose is genuinely impractical. The problem is the concentrated, fiber-free, liquid forms of fructose that define the modern food supply.
A 16-ounce soda contains the fructose equivalent of approximately 12 apples — minus the fiber, the volume, the chewing, and the satiety signals. A glass of orange juice delivers four oranges’ worth of fructose in 90 seconds. A handful of dried fruit can deliver more concentrated fructose than a full bowl of whole fruit, because dehydration concentrates the sugar while removing the water that limits intake.
These distinctions are not academic. They are mechanistic. The same total fructose grams delivered via whole fruit versus juice versus soda produce dramatically different liver fat outcomes — because the delivery vehicle determines whether the fructose hits the liver in a manageable trickle or a metabolic overload.
The ultra-processed food layer
Fructose alone explains a meaningful portion of the type 2 diabetes epidemic. But fructose is not consumed in isolation. It is consumed in the context of ultra-processed foods — and the combination amplifies the damage.
The peer-reviewed definition of ultra-processed foods is precise: “Ultra-processed foods (UPFs) are foods that have undergone extensive industrial processing with the addition of various substances in order to make them more tasty, eye-catching, and easy to consume. UPFs are usually rich in sugars, salt, and saturated fat, whereas they lack essential nutrients.”
The categories that fall under UPF:
- Packaged snacks (chips, crackers, snack cakes)
- Soft drinks and sweetened beverages
- Sugary breakfast cereals
- Industrial baked goods
- Frozen and ready-to-heat meals
- Reconstituted meat products (chicken nuggets, sausages, hot dogs)
- Most commercial breads
- Most commercial sauces, dressings, and condiments
- Sweetened yogurts and dairy desserts
- Most “low-fat” diet products (which substitute sugar for the removed fat)
The problem is not just the sugar content of UPFs, although that is real. The problem is the combination. UPFs are engineered with three additive features that drive metabolic disease together:
First, fructose density. UPFs are designed for palatability, and fructose is one of the cheapest ways to deliver palatability. The food industry’s discovery that high-fructose corn syrup could replace table sugar at a fraction of the cost transformed the American food supply in the 1970s and 1980s. By the time the metabolic consequences became clear, fructose was in everything.
Second, industrial seed oils. Soybean, corn, canola, sunflower, safflower, and cottonseed oils are nearly ubiquitous in UPFs. These oils provide the omega-6-rich substrate that gets oxidized into the inflammatory lipid byproducts that drive endothelial damage. The combination of high fructose and high omega-6 seed oil in the same food product is a metabolic double-hit.
Third, the chemical layer. Emulsifiers, artificial sweeteners, acrylamides, preservatives, food dyes, BPA leaching from packaging — these compounds have been independently linked to insulin resistance through inflammatory and gut microbiome pathways. None of them are individually catastrophic. The cumulative load from a diet that’s 60% UPF — which is roughly the modern American average — is.
The result is a food category that is designed for hyperpalatability, lacks the fiber and protein that would slow digestion, delivers concentrated fructose directly to the liver, provides the oxidative substrate that drives inflammation, and contains a chemical mix that further degrades metabolic function. Of course type 2 diabetes is exploding. The food supply has been engineered into a Twin Cycle accelerator.
The accelerants: sleep, stress, and sitting
The dietary mechanism is primary. But three lifestyle factors significantly accelerate the cascade, and any complete picture of T2D causation has to include them.
Sleep deprivation. One week of sleeping fewer than six hours per night produces measurable insulin resistance in healthy adults. Shift workers — people whose circadian rhythms are chronically disrupted — have approximately 40% higher type 2 diabetes risk than day workers, independent of diet and weight. The mechanism is multifactorial: sleep deprivation elevates cortisol, disrupts leptin and ghrelin signaling (driving overeating), impairs glucose tolerance directly, and reduces insulin sensitivity at the cellular level. You cannot out-diet chronic short sleep.
Chronic psychological stress. Cortisol — the body’s primary stress hormone — directly stimulates hepatic gluconeogenesis, the same process that’s already overactive in a fatty liver. Chronic stress effectively layers an additional insulin resistance signal on top of the dietary one. The patient who is stressed, undersleeping, and eating poorly is not three problems compounded — they are one problem reinforced through three independent channels.
Sedentary lifestyle. Skeletal muscle is the largest glucose disposal organ in the body. When muscles contract, they pull glucose out of the bloodstream — often through pathways that don’t require insulin at all. A sedentary lifestyle removes one of the body’s most important regulatory mechanisms. Worse, it allows muscle tissue to accumulate intramuscular fat — yet another ectopic deposition site that further drives insulin resistance. We’ll cover exercise as therapy in detail in Post 4. For now, the relevant point is that sitting is not just the absence of activity. It is an active driver of the disease.
These three accelerants do not cause T2D in the absence of the dietary driver. They amplify and accelerate it. A person consuming a clean diet but sleeping four hours, working a chronically stressful job, and not moving will eventually develop metabolic dysfunction. A person consuming UPF but sleeping eight hours, managing stress effectively, and lifting three times a week will develop it faster than the literature suggests they should. The lifestyle factors are not optional. They are part of the same metabolic system.
What the framework means
Once you understand the Twin Cycle, the entire architecture of mainstream type 2 diabetes care becomes visible as a category error.
The standard approach asks: how do we lower this patient’s blood glucose?
The mechanism asks: how do we get the fat out of this patient’s liver and pancreas so the disease resolves at the source?
These are not the same question. They lead to different treatments. They lead to different outcomes. And — as we’ll see in the next post — the pharmaceutical approach that focuses on glucose lowering can not only fail to address the underlying problem; in certain trial conditions, it has actually killed patients.
Roy Taylor’s summary, after fifteen years of imaging studies and intervention trials: “This means we can now see type 2 diabetes as a simple condition where the individual has accumulated more fat than they can cope with. Importantly this means that through diet and persistence, patients are able to lose the fat and potentially reverse their diabetes. The sooner this is done after diagnosis, the more likely it is that remission can be achieved.”
A simple condition. Not a complex multifactorial disease. A condition with one primary mechanism, two primary drivers, and three accelerants — all of which are addressable. The narrative of complexity that has surrounded T2D for thirty years is the narrative that keeps patients on lifelong medication. The simpler, mechanistic framework is the one that points toward resolution.
The drugs do not do that. That’s the next post.
What’s coming next
Post 3 takes on the pharmaceutical layer head-on. The ACCORD trial — what happens when you try to drive HbA1c down with medication. The GLP-1 / Ozempic phenomenon — what these drugs actually do, what they don’t do, and what happens when you stop. The honest accounting of metformin. The framework that explains why “lifelong management” was always the wrong framing.
The drugs are not the cure. They were never going to be. The mechanism is too specific, and the medications are too downstream.
We’re going there next.
This post is part of a four-part series on type 2 diabetes — what causes it, what your doctor isn’t telling you, what the drugs can and can’t do, and what actually reverses the disease.
- Pre-Diabetes Is Not the Early Stage of Type 2 Diabetes. It’s the Late Stage of Insulin Resistance.
- The Twin Cycle: What Actually Causes Type 2 Diabetes. (this post)
- The ACCORD Trial and the GLP-1 Trap: Why the Drugs Aren’t the Answer
- The Cure: How Type 2 Diabetes Is Reversed
